Orthogonal glycosylation


Orthogonal glycosylation

Chapter 4
Synthesis of extended bood type B determinant
Kanie, O.; Ito, Y.; Ogawa, T. Tetrahedron Lett., 1996, 37, 4551-4554.
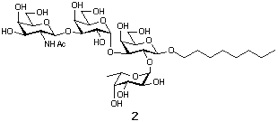
Sction 1: Synthetic Plan.
Execution of the synthesis used an orthogonal set of glycosylation reactions, namely activation of thioglycosides and glycosyl fluorides with conditions a (NIS-AgOTf [3] or DMTST [4] ) and conditions b (Cp2HfCl2-AgClO4 [5] ), respectively. Thus, four monosaccharide units 3, 4, 5, and 6 were synthesized as either glycosyl fluoride or thioglycoside to fulfill the criteria of this concept. All necessary selective protections as well as anomeric transformations were completed at the stage of monosaccharide, thereby, minimizing manipulations during elongation of the sugar chain.
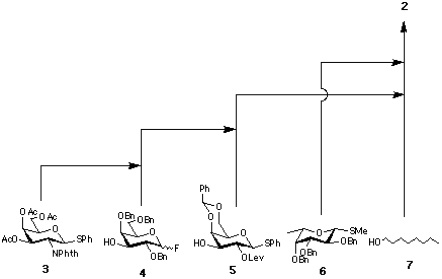
Section 2: Preparation of Monosaccharides.
Synthesis of 3
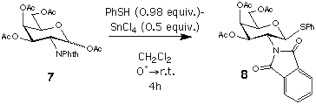
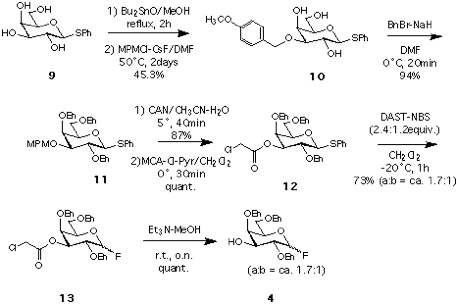
Synthesis of 4
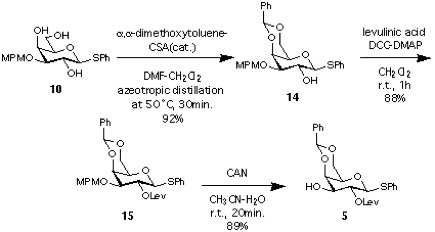
Synthesis of 5
Section 3: Synthesis of Extended Blood Type B Determinant
The synthesis was started from suitably protected galactosyl fluoride 4 (α:β = 1.7:1) which was glycosylated with GalNAc precursor 3 under conditions a (NIS-AgOTf) in CH2Cl2 (-20 to become 0 degree C) to afford 16 (90%). The stereochemical assignment was confirmed by 1H NMR, where J1',2' value of ca. 8.5Hz for H-1' indicated the β-D-configuration for GalNAc residue. No anomerization of the reducing end fluoride was observed during the glycosylation reaction which eliminated the potential ambiguity of this strategy. The disaccharide 16 was directly used as a donor for the next glycosylation reaction with the phenylthio glycoside 5. The glycosylation under conditions b in CH2Cl2-Et2O (-78 to become r.t.) in the presence of CaSO4 afforded desired trisaccharide 17 (73.4%). The a-configuration of the newly formed glycosidic linkage was evident from the coupling constant of the anomeric proton at δ 4.77 (J = 3.6Hz). Glycosylation of 17 with octyl alcohol 7 was then carried out under conditions a (NIS-AgOTf) in CH2Cl2 (-20 to become r.t.) to yield octyl glycoside 18 (89.7%). Deprotection of the levulinoyl group on trisaccharide 18 using hydrazine acetate [6] afforded 19 (85.4%) which was then used as acceptor for the α-fucosylation reaction. Thus, the levulinoyl group was used not only as a participating goup to yield α-glycoside but also as a selectively removable protecting group. The coupling reaction of 19 using methylthio glycoside 6 [7] (0 to become10 degree C) to give desired 20 (88%). Ethylenediamine [8] treatment of tetrasaccharide 20 followed by selective acetylation of the resulting amine yielded 2 (85%) after hydrogenolysis under acidic condition. Compound 18 was also deprotected to afford 21 in a same manner.
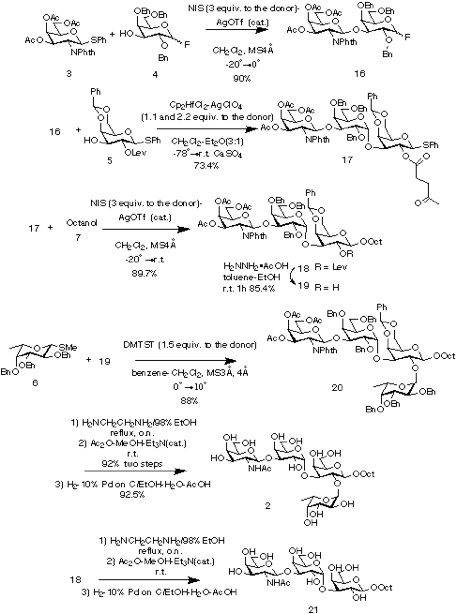
We demonstrated the applicability of the orthogonal glycosylation strategy to the synthesis of a branched oligosaccharide with four different types of glycosidic linkages. The synthesis of the target tetrasaccharide was thus achieved in seven steps (38% overall yield) from protected monosaccharide units. [9]
References
1. (a) Paulsen, H. Angew. Chem., Int. Ed. Engl., 1982, 21, 155-175. (b) Schmidt, R. R. Angew. Chem., Int. Ed. Engl., 1986, 25, 212-235. (c) Paulsen, H. Angew. Chem., Int. Ed. Engl., 1990, 29, 823-938. (d) Hindsgaul, O. Seminars in Cell Biology, 1991, 2, 319-326. (e) Wong, C.-H.; Halcomb, R. L.; Ichikawa, Y.; Kajimoto, T. Angew. Chem., Int. Ed. Engl. 1995, 34, 521-546. (f) Crawley, S. C.; Palcic, M. M. "Use of Glycosyltransferases in the Synthesis of Unnatural Oligosaccharide Analogues", in Modern Methods in Carbohydrate Synthesis, Khan, S. H.; O'Neill, R. A. Eds., Harwood Academic Publishers, United States, 1996, pp492-517.
2. Teneberg, S.; Jovall, P.-A.; Karlsson, H.; Sjogren, H.-O.; Brodin, T. J. Biochem. 1994, 116, 697-703.
3. (a) Konradsson, P.; Mootoo, D. R.; McDevitt, R. E.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun., 1990, 270-272. (b) Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett., 1990, 31, 1331-1334. (c) Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett., 1990, 31, 4313-4316.
4. Fugedi, P.; Garegg, P. J. Carbohydr. Res. 1986, 149, c9-c12.
5. (a) Suzuki, K.; Maeta,; H. Matsumoto, T.; Tsuchihashi, G. Tetrahedron Lett., 1988, 29, 3571-3574. (b) Suzuki, K.; Maeta, H.; Matsumoto, T. Tetrahedron Lett., 1989, 30, 4853-4856.
6. Koeners, H. J.; Verhoeven, J.; van Boom, J. H. Rec. Trav. Chim. Pays-Bas., 1981, 100, 65-72.
7. Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res., 1987, 167, 197-210.
8. Kanie, O.; Crawley, S. C.; Palcic, M. M.; Hindsgaul, O. Carbohydr. Res., 1993, 243, 139-164.


Chap. 1:
Chap. 2:
Chap. 3:
Chap. 4:
Chap. 5:
Chap. 6:




