Orthogonal glycosylation


Orthogonal glycosylation

Chapter 5
Application to polymer supported synthesis
Ito, Y., Kanie, O., and Ogawa, T. Angew. Chem. Int. Ed. 1996, 35, 2510-2512.
Chemical synthesis of glycoconjugate-related oligosaccharide requires a large number of manipulations, and inevitably becomes time-consuming. Since solid-phase synthesis has been demonstrated to be extremely valuable for routine preparation of oligopeptides and oligonucleotides, potential usefulness for the synthesis of other biomolecules is quite obvious. In addition, usefulness of polymer-support chemistry in combinatorial chemistry is also well recognized. From these points of view, oligosaccharide synthesis on polymer support is attracting recent attention[1-4]. We report here a novel strategy for polymer-support synthesis of oligosaccharides, which enables the preparation of biologically relevant oligosaccharides in a theoretically shortest way. Assembly of oligosaccharide backbone was performed on a polymer support based on the concept of orthogonal glycosylation strategy[5], and was followed by the incorporation of a hydrophobic aglycon as a tag enabling rapid extraction of desired product. According to this strategy, syntheses of tri- (13) and tetrasaccharide (18) were achieved in 40 and 30% overall yield, respectively.
Section 1: Introduction.
In polymer-support synthesis of oligosaccharide, two approaches can be considered with respect to the direction of chain elongation (Scheme); from reducing end to non-reducing end (Approach A), and from non-reducing end to reducing end (Approach B). Among them, the former approach is generally considered to be more advantageous because, as far as both polymer-supported aglycon and glycosyl donor are reactive enough to ensure completion of all glycosylations (step 1), and selective deprotection to liberate the hydroxy group for the next glycosylation (step 2) can be performed quantitatively, nearly homogeneous product can be obtained. On the other hand, the application of Approach B is less straightforward in principle, due to the considerations that follows. First, every glycosylation (step 1) inevitably gives rise to side reaction(s) (-elimination, hydrolysis, etc.) together with the formation of desired O-glycoside bond, and all products should be accumulated on the polymer supported phase. In addition, transformation of the reducing-end anomeric position into certain leaving group (step 2) is required after each glycosylation, which is by no means straightforward at oligosaccharide stage.
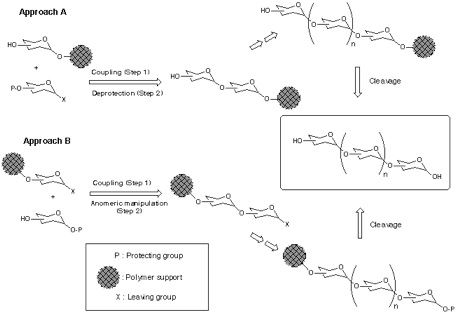
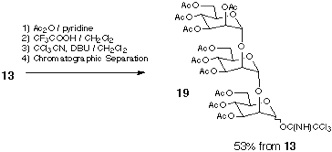
Addressing these problems inherent to Approach B, our attention has been turned into the application of orthogonal glycosylation strategy into polymer-support chemistry (Scheme). Combined use of thioglycoside and glycosyl fluoride was assumed, because these were demonstrated to be competent as a set of orthogonal glycosyl donors[5]. In order to avoid the potential difficulty of isolation of the final product, the assembly of oligosaccharide is to be followed by an incorporation of a hydrophobic aglycon as a tag at the reducing end anomeric position. Eventually, the target oligosaccharide can be distinguished from all other polymer-supported products, because it should become the only tag-carrying molecule. Therefore, the isolation process should be possible in a single step, irrespective of the number of glycosylation steps, by using reverse-phase silica gel-based technology[6]. In order to test this hypothesis, the first attempt was made using polyethylene glycol (PEG) as a "soluble" polymer[2], and the trimannoside portion (i.e. 13) of high-mannose type glycoprotein oligosaccharides[7] was chosen as a target. In this instance, 2-(trimethylsilyl) ethyl (SE) group was used as a hydrophobic tag[8].
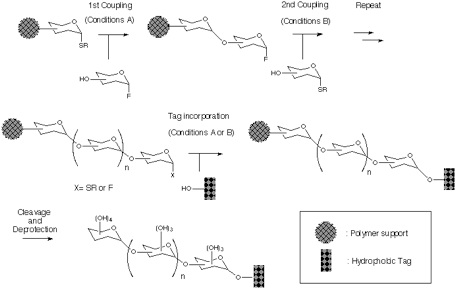
Section 2: Preparation of Polymer Supported Glycosyl Donor.
The synthesis was started from the ester 1, that was prepared from the chloride 2[9] via 3, 5, and 6. Saponification into acid 7, followed by coupling with PEG monomethyl ether (Aldrich, average MW 5,000) afforded the thiomannoside-polymer conjugate 8[10].
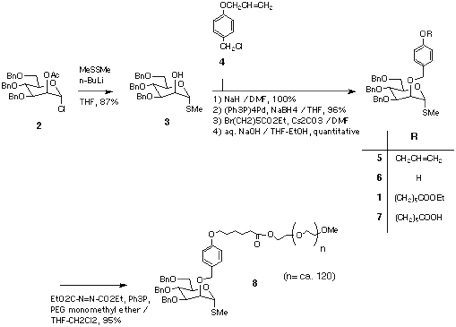
Section 3: Elongation of Sugar Chain.
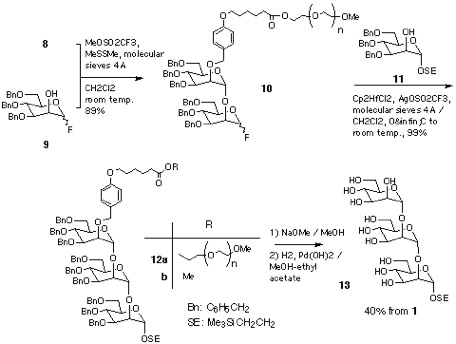
Section 4: Elongation of Sugar Chain-2(partial structure of GPI anchor).
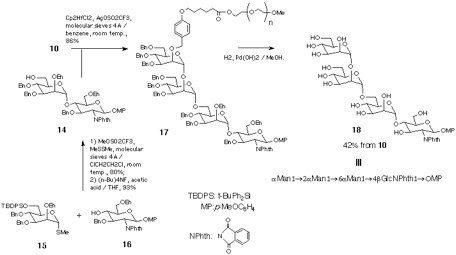
Disaccharide-PEG 10 was also converted into the tetrasaccharide 18 that corresponds to the partially protected form of the common carbohydrate structure of glycosyl phosphatidyl inositol (GPI) anchor[19]. Thus, the disaccharide 14 was synthesised from monosaccharide derivatives 15 and 16 and then reacted with 10, in the presence of Cp2HfCl2-AgOSO2CF3 in benzene[20]. Resultant tetrasaccharide-PEG conjugate 17 was subjected to catalytic hydrogenation. In this case, existence of p-methoxyphenyl and phthalimido groups makes the product hydrophobic enough to be retained in a C18 column. Thus, reverse-phase chromatographic separation, in a similar manner as described for 13, gave tetrasaccharide 18[17, 21] in 30% overall yield (Scheme 4).
References
1. Yan, L.; Taylor, C. M.; Goodnow Jr. R.; Kahne, D., J. Am. Chem. Soc. 1994, 116, 6953-6954 .
2. Douglas, S. P.; Whitfield, D.; Krepinsky, J. J., J. Am. Chem. Soc. 1994, 117, 2116-2117.
3. Schuster, M.; Wang, P.; Paulson, J. C.; Wong, C.-H., J. Am. Chem. Soc. 1994, 116, 1135-1136.
4. Randolph, J. T.; McClure, K. F.; Danishefsky, S. J., J. Am. Chem. Soc. 1995, 117, 5712-5719 .
5. Kanie, O.; Ito, Y.; Ogawa, T., J. Am. Chem. Soc. 1994, 116, 12073-12074.
6. Palcic, M. M.; Heeze, L. D.; Pierce, M.; Hindsgaul, O., Glycoconjugate J. 1988, 5, 49-63
7. Kobata, A., Acc. Chem. Res. 1993, 26, 319-324 .
8. For example; Ito, Y.; Paulson, J. C., J. Am. Chem. Soc. 1993, 115, 1603-1604. More hydrophobic aglycon similar to SE was reported recently; Stangier, P.; Palcic, M. M.; Bundle, D. R., Carbohydr. Res. 1995, 267, 153-159.
9. Ogawa, T.; Katano, K.; Sasajima, K.; Matsui, M., Tetrahedron, 1981, 37, 2779-2786.
10. All PEG-supported products were isolated by precipitation from t-butyl methyl ether as is described in ref. [2]. Yields were calculated based on weights and do no necessarily reflect the purity.
11. Ito, Y.; Ogawa, T., Angew. Chem. Int. Ed. Engl. 1994, 33, 1765-1767.
12. Lönn, H., J. Carbohydr. Chem. 1987, 6, 301-306.
13. Corresponding stereoisomer was not observed within the detection limit of 270 MHz 1H-NMR.
14. Prepared from fluoride 9b (1. Me3SiCH2CH2OH, Cp2HfCl2, AgOSO2CF3 / CH2Cl2: 2. NaOMe / MeOH).
15. The glycosylation can be performed in a nearly identical efficiency by using corresponding methylthioglycoside 3 as an acceptor.
16. Suzuki, K.; Maeta, H.; Matsumoto, T., Tetrahedron Lett. 1989, 30, 4853-4856.
17. 1H- (270 MHz) and 13C- (68.4 MHz)NMR data; 13: δH (D2O, 50 degree C) 5.25 (d, J 2 Hz, H-12), 5.06 (s, H-13), 5.03 (d, J 2 Hz, H-11), 4.09 and 4.05 (dd, J 3 and 2 Hz, H-21 and -22), 0.95 (m, CH2SiMe3), 0.02 (SiMe3); δC (D2O, 50 degree C) 103.68, 102.19, and 98.97 (anomeric carbons), 80.57 and 80.03 (C-21 and -22), 18.47(CH2SiMe3), -0.89 (SiMe3). 18: δH (D2O, 70 degree C) 5.70 (d, J 8.6 Hz, H-11), 5.28 (d, J 1.7 Hz, H-12), 5.13 (d, J 1 Hz, H-14), 5.06 (d, J 1.7 Hz, H-13); δC (D2O, 50 degree C) 103.64, 103.14, 99.88, and 98.83 (anomeric carbons). 19: CDCl3 δH 8.76 (NH), 6.43 (d, J 2.3 Hz, H-11), 5.20 (d, J 2.3 Hz, H-13), 4.97 (d, J 2.0 Hz, H-12).
18. This material was a 3.5:1 mixture of α- and β-anomers with respect to the Man2 residue. Isomers can be separated by eluting the C18 column with linear gradient of H2O to 50% MeOH.
19. McConville, M. J.; Ferguson, M. A. J., Biochem. J. 1993,294, 305-324.
20. Suzuki, K.; Maeta, H.; Suzuki, T.; Matsumoto, T., Tetrahedron Lett. 1989, 30, 6879-6882.
21. A 8:1 mixture of a- and b-anomers with respect to the Man2 was obtained. The reaction proceeded in a nearly stereorandom manner in CH2Cl2 (a:b=1.2:1).
22. Jansson, K.; Ahlfors, S.; Freid, T.; Kihlberg, J.; Magnusson, G., J. Org. Chem. 1988, 53, 5629-5647; Fukuyama, T.; Laird, A. A.; Hotchkiss, L. M., Tetrahedron Lett. 1985, 26, 6291-6292.
23. Schmidt, R. R.; Kinzy, W., Adv. Carbhydr. Chem. Biochem. 1994, 50, 21-123.


Chap. 1:
Chap. 2:
Chap. 3:
Chap. 4:
Chap. 5:
Chap. 6:




